
Cellular and Molecular Pharmacokinetics
Short description
We study how drug molecules are absorbed by the human body, how they distribute to different tissues, and how they are eliminated through metabolism and excretion.
We study how drug molecules are absorbed by the human body, how they distribute to different tissues, and how they are eliminated through metabolism and excretion. Our focus is on the cellular and molecular mechanisms involved, and we study these using a combination of biophysical and cell-based experiments, imaging, and computational modeling and simulation.
Of particular interest is the cellular disposition of non-traditional classes of drug molecules with challenging pharmacokinetics, including larger synthetic molecules such as macrocycles and targeted protein degraders (PROTACs), and biological molecules like oligonucleotide and peptide therapeutics.
Our research is centered around four interconnected themes: intracellular drug exposure, pharmacokinetics of non-traditional drug classes, membrane transporters and computational drug disposition modeling.
Intracellular drug exposure
We use computational and experimental methods to simulate, quantitate and visualize drug distribution at the tissue, cellular and subcellular scales.
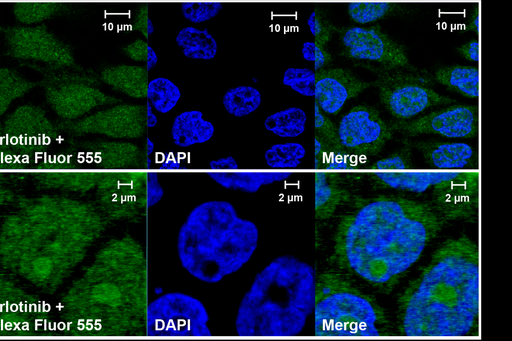
Most pharmacologically interesting proteins are located in the cell interior. Consequently, drug concentrations at intracellular or subcellular sites are driving the pharmacological and toxicological actions of the majority of drugs. Together with the Per Artursson lab at Uppsala University we have developed an assay that allows mass spectrometric measurements of unbound intracellular drug concentrations, i.e., the drug concentration that is available to interact with intracellular targets. We are now exploring the use of this methodology for new classes of drug molecules, in combination with other biophysical measurements, imaging techniques and computational modeling and simulation, to understand the mechanisms of intracellular drug distribution and the connection to cellular pharmacology.
Pharmacokinetics of non-traditional drug classes
We investigate how new therapeutic modalities can be designed to reach their intended targets in the body.
Small molecules (<500 Da) have conventionally been favored in drug development, due to a relatively simpler optimization of their pharmacokinetic properties, and because they are suited for of the types of pharmacological targets that have historically been pursued. However, many interesting target types are difficult to modulate with small molecule ligands, and non-traditional chemotypes are therefore increasingly explored. Prominent examples include macrocyclic molecules, targeted protein degraders (PROTACs), and RNA-based therapeutics, that could bring new regions of the human proteome into reach for drug development.
Currently, clinical progress is hampered by that the molecular properties of these new therapeutic modalities typically are incompatible with oral absorption and intracellular exposure. Using combinations of cell-based in vitro experiments, biophysical measurements and computational modeling, we study how such non-traditional drugs can be designed to cross membrane barriers and reach their sites of action.
Membrane transporters in drug disposition
We study the role of membrane transporters in the absorption, cellular uptake, tissue distribution and elimination of drug molecules. A particular focus is on the molecular mechanisms of transporter-mediated drug interactions and -translocation.
Hundreds of distinct proteins mediate transport of small-molecule solutes across biological membranes. These include the members of the ATP-Binding Cassette (ABC) and Solute Carrier (SLC) protein superfamilies, of which currently a couple of dozens have demonstrated roles in the disposition of drug molecules.
Many questions concerning drug transporters remain unanswered, including which of all transporters are relevant for drug-like molecules, how these are expressed, regulated and dynamically presented in human tissues, and what the molecular properties of drug molecules are that lead to their binding to the transporter, their translocation, and/or to transporter mediated drug–drug interactions? We use a combination of in vitro measurements and computational modeling techniques to address such questions.
Computational drug disposition modeling
We use computational modeling and simulation to shed light on the cellular and molecular mechanisms of drug disposition, and to develop predictive tools for improved, more rational drug development.

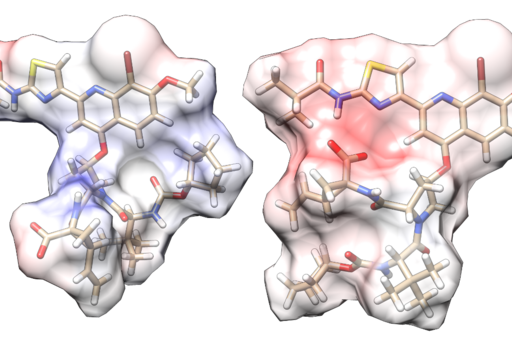
Modeling is performed at multiple scales using a wide range of techniques. We use a combination of physics-based approaches (molecular dynamics, docking) and empirical structure-property relationship (QSPR) and protein structure modeling to understand the molecular interactions between drugs and cellular membranes and the function of transport proteins.
At the cellular and tissue levels, kinetic and physics-based (stochastic dynamics) techniques are used to study the impact of and interaction between different drug distribution phenomena, and physiologically-based pharmacokinetic modeling is used to connect to the in vivo clinical setting. At all levels data from experiments are integrated in the modeling, to improve predictions and to rationalize experimental observations.
Structure-property relationship models relate descriptions of the molecular structures of drugs to a measured property. The models can then be used to forecast properties of new molecules based solely on their molecular structure. We use such techniques to develop predictive models of, for example, cell permeability and drug interactions with transport proteins, through the use of a variety of machine learning techniques (e.g., partial least squares, random forests and support vector machines).
In a molecular docking experiment, the interactions between molecules (e.g., a drug molecule and a pharmacological target protein or a transporter) are quantified and used to predict the three-dimensional orientation of the molecules that result in optimal binding.
Molecular dynamics simulations are used to describe the motion and interaction of molecules at atomic resolution. We use molecular dynamics to, for example, quantify and visualize the transport of drug molecules across biological membranes. For simulations at greater scales, such as whole cells and multicellular tissues, we use stochastic dynamics to simulate drug distribution phenomena.
Physiologically based pharmacokinetic (PBPK) modeling is used to model and simulate drug absorption, distribution, metabolism and excretion at the organ or whole-body level, using physiologically relevant parameters (for example, transport and metabolic rates, blood flows and organ volumes). Our research into the cellular and molecular mechanisms of drug absorption and disposition feed into such models, and our data has been implemented in the major software platforms for PBPK modeling.
Group members
Pär Matsson
Professor, Principal Investigator
Emma Inganäs
PhD student
Goran Khodayari
Diploma project (Pharmacist Program)
Anna Nyback
Research Internship Medicine


