
Kraniosynostos – kirurgi, genetik och neuropsykologi
Kort beskrivning
Projektets långsiktiga mål är att förbättra resultaten av kirurgisk behandling och därigenom livskvaliteten för de barn som drabbas av kraniosynostos, dvs. för tidig sammanväxning av skallbenets suturer. Vidare syftar projektet till att öka kunskaperna om de bakomliggande genetiska förändringar som ger upphov till kraniosynostos.
Projektet ingår i ämnesområdet plastikkirurgi och har tre huvudspår:
- Systematisk utvärdering av kirurgisk behandling och utveckling av nya operationsmetoder för patienter med kraniosynostos.
- Genetiska analyser, främst mutationscreening, av patienter med kraniosynostoser.
- Långtidsuppföljning av patienternas intellektuella och psykosociala funktionsförmåga.
Projektet är således multidisciplinärt för att kunna belysa så många aspekter som möjligt av kraniosynostsos och dess behandling.
Bakgrund
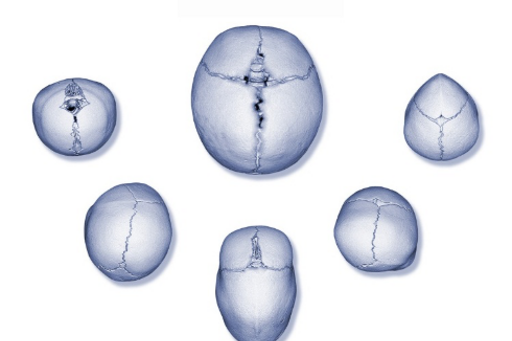
Det nyfödda barnets skalle växer efter förlossningen mycket snabbt. Under det första levnadsåret tredubblar hjärnan sin volym vilket förutsätter att neurokraniet är eftergivligt och tillåter tillväxt. Under det första levnadsåret är skallens suturer effektiva och står för det mesta av skallens tillväxt. Prematur kraniofacial synostos innebär att suturerna slutits för tidigt, i regel intrauterint. Beroende på vilken av suturerna som slutits för tidigt uppstår karaktäristiska deformiteter av skallen. Vanligtvis är en enstaka sutur för tidigt sluten men synostos av flera suturer samtidigt, och som del av ett mer komplext syndrom, förekommer (Fig 1).
Incidensen av kraniofacial synostos är 1 barn per 1500 - 2000 födda, dvs. ca 50 - 75 svenska barn per år. Kraniofaciala enheten vid Plastikkirurgiska kliniken, Sahlgrenska Universitetssjukhuset har sedan 2012 tillstånd att bedriva Nationell Högspecialiserad vård (tidigare Rikssjukvård) inom detta område. Majoriteten av patienterna med kraniosynostos i Sverige handläggs på Sahlgrenska.
Kraniosynostos kan leda till allvarlig störning av hjärnans normala utveckling och en drastiskt försämrad livskvalitet, både för det drabbade barnet och dess familj. I de mest komplexa syndromala fallen gör ökat intrakraniellt tryck att operation är nödvändigt för att undvika blindhet och i värsta fall död. Även i fall av isolerad kraniosynostos finns mycket som talar för att utfallet av kirurgi är viktigt för barnens utveckling. Det finns fortfarande en stor lucka i vår kunskap om det långsiktiga utfallet av tidigare gjorda ingrepp, vilken behöver fyllas innan riktlinjerna för behandling kan göras evidensbaserade och optimerade.
Kirurgi
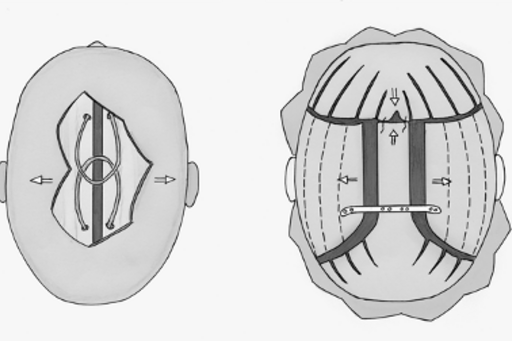
För 15 år sedan introducerades en ny operationsmetod av det kraniofaciala teamet i Göteborg. Dynamiska fjädrar i rostfritt stål implanterades för att påverka det växande kraniets form. Hittills har mer än 200 barn opererats med denna typ av fjädrar i Göteborg. Fjädrarna tolereras mycket väl och får sitta på plats i ca 6 månader varefter de tas bort med ett enkelt ingrepp. Operation med fjädrar är avsevärt enklare än en stor kranioplastik för barnet men ger i många fall ett lika bra resultat som mer omfattande kranioplastik (Fig 2). Blodförlusten i samband med ingreppet är minimal, morfinbehovet lågt, återhämtningen snabb och sjukhusvistelsen kort. Metoden är lätt att lära och sprids till flera centra vilket gör att fler patienter kan komma i fråga för denna operation. Fjädrarna har störst betydelse vid operation för sagittal synostos men används även vid metopikasynostsos, unikoronar synostos och för bakre skallexpansion.
De senaste resultaten:
Fjädertekniken och distraktorteknik har införts vid operation för unikoronar synostos. Hittills har 5 fall opererats och vid 6 månader efter operationen har ansiktsskoliosen upphävts helt, dvs barnen har blivit helt raka och symmetriska i ansiktet. Tidigare korrigerades enbart den tillbakasatta pannan medan skallbasen och ansiktet förblev lika skevt. Detta är en stor förbättring för patienter med unikoronar synostos.
En stor genomgång av alla kraniofaciala operationer under 10 år (1023 operationer) har gjorts och samtliga komplikationer har kartlagts och klassificerats enligt tre olika system. Det system som fungerar bäst (Oxfordskalan) har införts i klinisk rutin from 1 januari 2020 och samtliga komplikationer registreras nu enligt detta system. Det innebär att vi kan jämföra komplikationspanorama mellan olika kraniofaciala centra på ett objektivt sätt. Vår teknik med fjädrar vid sagittal synostos uppvisade en mycket gynnsam komplikationsprofil.
Vi har visat att tre fjädrar inte bidrar mer än marginellt jämfört med två fjädrar vid sagittal synostos. Effekten blir initialt lite kraftigare med större breddning av huvudet men vid tre års ålder, är effekten lika god oavsett om två eller tre fjädrar använts. Det innebär att vi kan nöja oss med en lite mindre omfattande operation i flertalet fall.
Arbetet med att finna nya användningsområden för de skonsamma fjädrarna har resulterat i att vi visat att de fungerar utmärkt som en första operation vid komplicerad, multipel synostsos. Då är det trångt i skallen och lillhjärnan tenderar att tränga ner i Foramen Magnum (Arnold Chiari). Multipla osteotomier och multipla fjädrar har visat sig kunna korrigera både form, volym och tendensen till "Arnold Chiari". Detta ger i flera fall tillräcklig effekt för att slippa ytterligare operationer men kan också vara en värdefull, lindrig första operation så att barnen hinner växa till sig innan nästa operation.
En genomgång av fyra års osteotomier kombinerat med fjädrar visar att det i nästan 20 % av fallen uppstår en ny radiologiskt till synes normal sutur på platsen där den normalt skulle ha bildats intrauterint. Fyndet är överraskande och antyder att komplicerade biologiska mekanismer spelar roll omkring vecka 20 i fosterlivet, då synostosen bildas. Möjligen kan det vara så att osteotomin och fjädrarna möjliggör för lokala signaler att skapa suturen. Denna upptäckt kan utgöra grunden för ett mer cellbiologiskt inriktat projekt om suturernas biologi.
Bakre skallexpansion är ett ingrepp som görs när trycket i skallen är högt. Vid komplexa synostoser och i syndromfall är det vanligt att man behöver operera flera gånger under de första levnadsåren för att ge hjärnan tillräckligt utrymme. Vi har utvecklat en metod med en bakre cirkumferent osteotomi som avlöser hela bakhuvudet men låter benet sitta kvar på hårda hjärnhinnan. Multipla fjädrar runt osteotomin pressar bakhuvudet bakåt och ökar volymen på ett relativt skonsamt sätt. Alldeles nya data visar att volymökningen är ca 200 ml och vi får momentan regress av huvudvärk och svullnad av synnervsutträdet i ögat.
I ett samarbete med UCL (University College London) (Biomechanical Engineering, Mehran Moazen PhD), tar vi vår fjäderteknik vid sagittal syntostas till ett biomekaniskt projekt. Det går ut på att isolera alla de dimensionsförändringar som sker i kraniet som svar på operationen. Den data som genereras kommer i sin tur att ligga till grund för simuleringar av hur en skalle kommer att förändras som svar på olika typer av kirurgi.
Schematisk bild av de två operationstekniker vi använder vid sagittal synostos
Ett vårdvetenskapligt projekt har visat att genom att avstå det morfindropp som varit rutin första natten efter operationen så kan tiden på CIVA minskas från ett dygn till 6 timmars observation för barn som opereras med kraniotomi och fjädrar, vilka utgör majoriteten av kraniosynostosfallen. Barnen mår bättre och börjar äta tidigare. Förutsättningen för denna förändring är att inte säkerheten försämras och genomgång av samtliga opererade fall visade att inte i något fall under 20 års tid behövdes reoperation under första dygnet, dvs efter 6 timmars observation kan den fortsatta vården ske på vanlig vårdavdelning. Förutom att patienterna mår bättre och kan vårdas i en lugnare miljö så sparas CIVA-resurser.
Genetik
Mutationer i 12 gener leder till syndromala tillstånd med kraniosynostos som huvudsymtom: FGFR1 (Fibroblast growth factor receptor 1), FGFR2 (Fibroblast growth factor receptor 2), FGFR3 (Fibroblast growth factor receptor 3), TWIST1 (Twist homolog Drosophila 1), EFNB1 (Ephrin-B1), RAB23 (Ras-associated protein), MSX2 (Muscle segment homeobox Drosophila 2), TGFBR1 (Transforming growth factor-ß receptor typ 1), TGFBR2 (Transforming growth factor-ß receptor typ 2), POR (Cytochrome P450 oxidoreduktas), FBN (Fibrillin) och TCF12 (Transcription factor 12). Utöver dessa gener finns ett stort antal gener med svagare koppling till kraniosynostos.
Genetiska studier har en direkt klinisk tillämpning i och med att ökad kunskap om de kända genetiska förändringarna som orsakar ärftliga kraniofaciala syndrom bidrar till en förbättrad förståelse av den kliniska fenotypen och prognosen samt ger möjlighet till en anpassad uppskattning av upprepningsrisken i familjen (genetisk vägledning).
De senaste resultaten:
DNA från 144 av 150 patienter har uppfyllt kvalitetskravet för att kunna genomgå Next Generation Sequencing-analys. Hos 89 av 144 (ca 62%) analyserade patienter har vi funnit kända patogena eller sannolikt patogena genvarianter som kan förklara patienternas kliniska bild. Majoriteten av varianterna återfanns i ”klassiska” kraniosynostosgener, dvs FGFR2, TWIST1, FGFR3, FGFR1, TCF12, EFNB1 och POR.
Vi har kunnat påvisa nya tidigare icke-beskrivna varianter i FGFR2-genen (2 nya varianter), FGFR1 (1 variant), TWIST1 (3 varianter), TCF12 (3 varianter) och POR (1 variant). Dessa varianter uppfyller kriterierna för att klassas som sannolikt patogena och är fenotypiskt korrelerade.
Förutom patogena varianter i tidigare kända gener med koppling till kraniosynostos, har vi funnit patogena och sannolikt patogena varianter i 2 gener (2 patienter) vars mutationer leder till syndromala tillstånd med dokumenterad kraniosynostos: 1 ny sannolik patogen variant i KMT2D-genen hos en patient med kliniskt Kabuki syndrom typ 1 (OMIM# 147920) och 1 tidigare beskriven patogen variant i SKI-genen som förknippas med Shprintzen-Golberg syndrom (OMIM# 182212).
Utöver det har vi påvisat 2 nya varianter i homozygot form i IL11RA-genen (2 patienter) som förknippas med kraniosynostos och tandavvikelser (OMIM# 614188). IL11RA-genen kan vara central för dessa komplexa tillstånd och ska därmed inkluderas i en mer omfattande genpanel för kraniosynostos. Denna kunskap är ett exempel på hur forskningen förbättrar den kliniska genetiska analysrutinen.
Hos en patient har vi funnit en kombination av två genetiska förändringar som båda uppstått de novo (sporadiskt), dels en Kabuki syndrom associerad trunkerande variant i KMT2D-genen samt en 3,2 Mbp 10q22.3q23.1 mikrodeletion som detekterats med hjälp av SNP-array analys innan den aktuella mutationsscreeningen utfördes. Detta ovanliga fall illustrerar betydelsen av en noggrann klinisk bedömning av patienterna med kraniosynostos inför tolkningen av genetiska resultat samt styrker att kraniosynostos är en del av den kliniska bilden vid Kabuki syndrom.
Man kan anta att det finns ett mörkertal av patienter med mer än en patogen genetisk förändring som kan förklara komplexa fenotyper där kraniosynostos är en del av den kliniska bilden tillsammans med andra missbildningar.
Hos 33 patienter av 144 analyserade (ca 23%) har vi inte funnit någon patogen eller sannolikt patogen variant i någon av de 63 sekvenserade generna. Däremot har vi i dessa fall noterat varianter av idag okänd klinisk signifikans (VOUS– variant of unknown clinical significance) i ett flertal gener i panelen. Här behövs fördjupad bedömning av såväl genotyp som patientens fenotyp för att dessa varianters eventuella sjukdomsorsakande effekt skall kunna fastställas.
Hos 22 av 144 analyserade patienter (ca 15%) har vi inte funnit någon genetisk avvikelse alls.
Sammantaget har vår inledande studie visat att en bred genetisk screening av kraniosynostos-patienter har en hög träffsäkerhet (ca 62%) och ger samtidigt möjlighet att förklara etiologin vid komplexa fenotyper som ej tillhör de klassiska kraniosynostossyndromen. Studierna ger också unika möjligheter att identifiera nya gener och mutationer som orsakar kraniosynostos.
Neuropsykologi
Då kraniofacial kirurgi bedrivits vid plastikkirurgiska kliniken sedan drygt 40 år finns möjlighet att göra långtidsuppföljning av både det kirurgiska resultatet men också av hur den neuropsykologiska utvecklingen, livssituationen och livskvaliteten utvecklas ända upp i vuxen ålder för dessa patienter.
Vi har tidigare genomfört ett par enkätbaserade studier för två av de syndromala grupperna.
För patienter med Apert syndrom kunde vi visa att flertalet hade någon form av utbildning och sysselsättning men att deras stora sociala problem var avsaknad av socialt nätverk. De bodde ofta kvar hemma hos sina föräldrar och hade få vänner.
För patienter med Crouzon syndrom var situationen i vuxen ålder generellt bättre. Spannet i livssituation var stort och flertalet levde ett relativt fullödigt liv med utbildning, jobb och familj.
Bland patienter med isolerad synostos har flera studier kunnat visa på ökad förekomst av störningar i neuropsykologisk utveckling. Även förekomst av t.ex. koncentrationssvårigheter och inlärningssvårigheter verkar vara särskilt vanligt vid metopikasynostos, men förekommer även vid sagittal synostos. Studier har visat att operation för sagittal synostos gör att barnen återhämtar den försening i tidig utveckling som kunde ses, samtidigt som de barn med sagittal synostos som inte blev opererade inte hämtade ifatt utvecklingsmässigt. Effekten var märkbar under lång tid. Andra studier har hävdat att ju större kirurgi som utförs och ju tidigare den görs desto mer gynnsamt är detta ur utvecklingssynpunkt. Dessa studier har fått stort genomslag men har metodologiska svagheter.
De senaste resultaten:
Vi har mätt fullskale-IQ (WAIS-IV från 16 års ålder och WISC-IV för 6 – 16åringar) hos barn som opererats för kraniosynostos. Vårt bortfall är unikt litet, vilket kan bero på att vi kallat patienter boende i Göteborg och sedan vidgat materialet stegvis genom att kalla patienter som bor längre och längre bort från Sahlgrenska Universitetssjukhuset. Vi undviker då bortfall pga alltför lång resväg. Preliminära resultat indikerar på helt normal IQ för isolerad synostos och vi har ett medelvärde och en spridning av värdena som är kongruent med normalfördelningen. Detta skiljer sig från flera publikationer i fältet där man istället ser medelvärden för fullskaligt IQ som är mycket höga, upp till 112, vilket ter sig helt orimligt. Endera har man i dessa studier en stor selektionsbias eller så använder man instrumenten felaktigt. Våra resultat tyder på att vi använder instrumenten korrekt och att vår strategi med urval av patienter med kort avstånd till Sahlgrenska Universitetssjukhuset ger unikt låg selektionsbias.
Ett specifikt test av uppmärksamhet (Conners CPT) har visat flera mindre avvikelser från normdata.
Allmänna förmågor har undersökts genom rapport från föräldrar (ABAS) och även här finner vi mindre avvikelser.
Sammantaget visar dessa studier att ungdomar som opererats för sagittal synostos och metopikasynostos har en normal utveckling men att flera små avvikelser från normen kan identifieras.