
Craniosynostosis: surgery, genetics and neuropsychology
Short description
The long-term goal of the project is to improve the results of surgical treatment, and thereby quality of life, for children affected by craniosynostosis, a condition in which the skull-bone sutures (junctions) fuse prematurely. A further project aim is to learn more about the genetic mutations underlying craniosynostosis.
The project comprises three main lines of research:
- Systematically reviewing surgical treatment and developing new procedures for operating on patients with craniosynostosis.
- Performing genetic analyses, especially mutation screening, of patients with craniosynostoses.
- Long-term monitoring of patients’ intellectual and psychosocial functional capacity.
For maximum clarification of craniosynostosis and its treatment, the project is multidisciplinary.
Background
The newborn baby’s skull grows very fast. In the first year of life, brain volume triples. The neurocranium must be flexible to allow this growth. Up to the age of one year, the sutures (gaps between skull bones) are effective, and largely account for skull growth. Premature craniofacial synostosis means that the sutures close too early, usually in utero. Depending on which of the sutures fuses prematurely, characteristic deformities of the skull occur. Usually, premature closure of only one suture at a time takes place, but synostoses of several sutures simultaneously may occur as part of a more complex syndrome (Figure 1).
The incidence of craniofacial synostosis is 1 child per 1500–2000 born, i.e. some 50–75 Swedish children annually. Since 2012, the Craniofacial Unit at the Department of Plastic Surgery, Sahlgrenska University Hospital has been licensed to conduct what is known as “national highly specialized care” (formerly “national health care”) in this area. Most patients with craniosynostosis in Sweden are treated at Sahlgrenska.
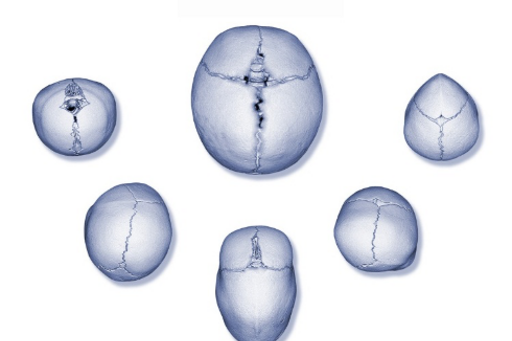
Figure 1. The central image shows a normal skull with open sutures and an oval head shape. Around it, from left to right, are skulls with bicoronal, unicoronal, sagittal, lambdoid, and metopic synostoses respectively, each with its own characteristic shape change.
Craniosynostosis can lead to severe disruption of normal brain development and drastically reduced quality of life, both for the children affected and for their families. In the most complex syndromic cases, increased intracranial pressure necessitates surgery to avoid blindness and, at worst, death. Even in cases of isolated craniosynostosis, there is substantial evidence that the outcome of surgery has a major bearing on these children's development. In our knowledge of the long-term outcome of procedures performed in the past, there is a large gap that needs to be filled before treatment guidelines can be made evidence-based and optimized.
Surgery
A new surgical method was introduced by the craniofacial surgery team in Gothenburg 15 years ago. They began to implant dynamic stainless-steel springs to affect the shape of the growing skull. To date, more than 200 children have had springs of this type surgically inserted in Gothenburg. The springs are very well tolerated and can stay in place for about 6 months, after which they are removed in a simple procedure. Surgical spring insertion is considerably simpler than performing a large cranioplasty (cranial reconstruction) for the child, and in many cases the former’s results are also just as good as those of more extensive cranioplasty (Figure 2). Blood loss in the course of the procedure is minimal, morphine requirements are low, recovery is fast and the hospital stay is brief. The method is easy to learn and is spreading to several centers, which means that more patients can be considered for this operation. The springs are most important in surgery for sagittal synostosis, but they are also used in metopic and unicoronal synostosis, and for posterior skull expansion.
Techniques using springs and distractors (devices that slowly move the cranial bones apart) have been introduced in surgery for unicoronal synostosis. To date, operations of this kind have been performed in 5 cases. At 6 months after the operation, the facial scoliosis has been entirely eliminated, i.e. the children’s faces have become completely straight and symmetrical. In the past, only the retracted forehead was corrected while the skull base and face remained equally lopsided. The new techniques thus represent a great improvement for patients with unicoronal synostosis.
A major review of all (1023) craniofacial operations in a 10-year period has been carried out, and all complications have been mapped and classified according to three different systems. The system that works best (the Oxford Scale) was introduced into routine clinical practice from 1 January 2020, and all complications are now registered in line with this system. This enables us to objectively compare different craniofacial surgery centers in terms of the whole range of complications. Our technique of using springs for sagittal synostosis has shown a highly favorable complication profile.
We have shown that, in sagittal synostosis, three springs contribute only marginally more than two. The effect is initially a little stronger, with greater widening of the head, but by the age of three years the effect is equally good irrespective of whether two or three springs are used. In most cases, we can thus confine ourselves to a slightly less invasive procedure.
Our work to find new application areas for the gentle springs has culminated in our demonstration that they work excellently in the patient’s first operation for complicated, multiple synostosis. There is then a shortage of space in the skull, and the cerebellum tends to penetrate into the foramen magnum (an “Arnold–Chiari” malformation). Multiple osteotomies and multiple springs have been shown to correct shape, volume and Arnold–Chiari tendency alike. In several cases, this is sufficiently effective for further surgery to be avoided, but it can also be a valuable, mild initial operation that gives these children time to grow before the next operation.
A review of four years’ osteotomies combined with springs shows that in almost 20% of cases a new suture that, radiologically, appears to be normal arises at the site where it would normally have formed in utero. This finding is surprising, and indicates that there are complicated biological mechanisms at work in about week 20 of gestation, when the synostosis is formed. It is possible that the osteotomy and the springs enable local signals to create the suture. This discovery can serve as the basis for a project focusing more on cell biology, and specifically on the biology of sutures.
Posterior skull expansion is a procedure performed when there is excessive pressure in the skull. In complex synostoses and syndrome cases, it is common for several operations to be necessary during the child’s first years of life, to give the brain enough space. We have developed a method involving an osteotomy around the posterior circumference, replacing the entire back of the head but leaving the bone on the dura mater (the hard outer layer of the meninges). Multiple springs around the osteotomy push the back of the head backward and increase the volume in a relatively gentle way. Brand-new data show that the volume is enlarged by about 200 ml, and we obtain instant regression of headache and of swelling of the optic nerve exit in the eye.
In a collaboration with University College London (Biomechanical Engineering, Mehran Moazen PhD), we are using our spring technique in sagittal synostosis in a biomechanical project. This involves isolating all the dimensional changes that occur in the skull in response to the operation. The data generated will, in turn, pave the way for simulations of how a skull will change in response to different types of surgery.
Schematic illustration of the two surgical techniques we use in sagittal synostosis
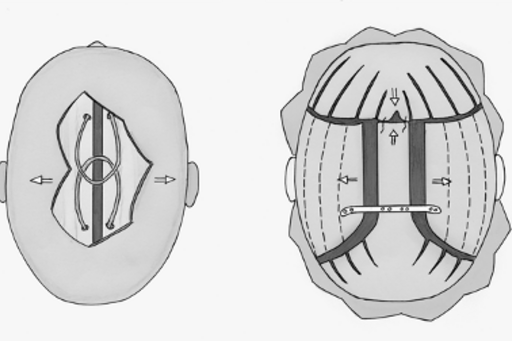
Figure 2. The image on the left shows the spring technique with reduced brain exposure, smaller bone-sawing incision area and two springs pushing the parietal bones apart. To the right, use of polyimide (PI) plastic is shown, with a considerably greater area of skull and dura mater removed and very extensive sawing in the skull.
A care science project has shown that withholding the morphine drip that used to be routine during the patient’s first night after the operation can reduce the required observation period in Sahlgrenska’s central intensive care unit (CIVA) from 24 to 6 hours for children who undergo craniotomy with spring insertion, who make up the majority of craniosynostosis cases. Postoperatively, these children feel better and start eating earlier. The prerequisite for this change is that there is no decrease in safety. A review of all surgical cases showed that not in a single case in 20 years was a repeat operation needed during the 24 hours — that is, after 6 hours’ observation further care can be provided in a regular ward. Besides the patients feeling better and being able to receive care in a more peaceful environment, CIVA resources are saved.
Genetics
Mutations in 12 genes lead to syndromal conditions with craniosynostosis as the main symptom: FGFR1 (fibroblast growth factor receptor 1), FGFR2 (fibroblast growth factor receptor 2), FGFR3 (fibroblast growth factor receptor 3), TWIST1 (twist homolog [Drosophila] 1), EFNB1 (Ephrin-B1), RAB23 (Ras-related protein), MSX2 (muscle segment homeobox [Drosophila] 2), TGFBR1 (transforming growth factor-ß receptor type 1), TGFBR2 (transforming growth factor-ß receptor type 2), POR (cytochrome P450 oxidoreductase), FBN (fibrillin), and TCF12 (transcription factor 12). Besides these genes, there are numerous others that are more loosely connected with craniosynostosis.
Genetic studies have direct clinical applicability since greater knowledge of the known genetic changes that cause hereditary craniofacial syndromes helps to improve our understanding of the clinical phenotype and prognosis, and provides an opportunity for individualized assessment of the risk of recurrence in the family (genetic counselling).
Latest results
DNA from 144 out of 150 patients met the quality requirement for undergoing next-generation sequencing analysis. In 89 of the 144 (approximately 62%) patients analyzed, we found known pathogenic, or probably pathogenic, gene variants that can explain the patient’s clinical symptoms. The majority of these variants were found in “classical” craniosynostosis genes, i.e. FGFR2, TWIST1, FGFR3, FGFR1, TCF12, EFNB1, and POR.
We have been able to establish new, previously undescribed variants in the FGFR2 gene (2 new variants), FGFR1 (1 variant), TWIST1 (3 variants), TCF12 (3 variants), and POR (1 variant). These variants meet the criteria for classification as probably pathogenic, and are phenotypically correlated.
Besides pathogenic variants in previously known genes linked to craniosynostosis, we have found pathogenic and probably pathogenic variants in 2 genes (2 patients) whose mutations lead to syndromal conditions with documented craniosynostosis. These were: 1 new probable pathogenic variant in the KMT2D gene, in a patient with clinical Kabuki syndrome type 1 (OMIM [Online Mendelian Inheritance in Man®] # 147920), and 1 previously described pathogenic variant in the SKI gene associated with Shprintzen – Goldberg syndrome (OMIM # 182212).
In addition, in the IL11RA gene (2 patients) we have identified 2 new variants in homozygous form that are associated with craniosynostosis and dental abnormalities (OMIM # 614188). The IL11RA gene may be central to these complex conditions and should therefore be included in a more comprehensive gene panel for craniosynostosis. This knowledge is an example of how research improves the clinical routine for genetic analysis.
In one patient, we found a combination of two genetic changes that both occurred de novo (sporadically): a truncating variant, associated with Kabuki syndrome, in the KMT2D gene and a 3.2 Mbp 10q22.3q23.1 microdeletion, detected by using SNP (single nucleotide polymorphism) array analysis before the mutation screening concerned was performed. This unusual case illustrates the importance of meticulous clinical assessment of patients with craniosynostosis before interpretation of the genetic results, and provides evidence that craniosynostosis is part of the clinical picture in Kabuki syndrome.
In 33 patients out of 144 analyzed (approximately 23%), we have not found any pathogenic, or probably pathogenic, variant in any of the 63 genes sequenced. On the other hand, in these cases we have noted, in a number of genes in the panel, variants of currently unknown clinical significance (VOUS: variant of unknown clinical significance). An in-depth assessment of both the genotype and the patient's phenotype is needed here, in order for the possible disease-causing effect of these variants to be determined.
In 22 out of 144 patients (approximately 15%) analyzed, we found no genetic abnormality at all.
Overall, our initial study showed that a broad genetic screening of craniosynostosis patients has high accuracy (approximately 62%) and simultaneously provides means of explaining the etiology of complex phenotypes that are not among the classic craniosynostosis syndromes. The studies also give unique opportunities to identify new genes and mutations that cause craniosynostosis.
Neuropsychology
Craniofacial surgery has been performed at the Department of Plastic Surgery for over 40 years. This has made possible long-term monitoring of not only these patients’ surgical outcomes, but also of how their neuropsychological condition, life situation and quality of life develop right up to adulthood. We have previously conducted a couple of questionnaire surveys of two of the syndromal groups. For patients with Apert syndrome, we were able to show that the majority had some form of education and employment, but that their major social problem was the lack of a social network. They were often still living at home with their parents and had few friends. For patients with Crouzon syndrome, the situation in adulthood was generally better. The range of life situations was large, but most lived a relatively full life with education, work and family.
Among patients with isolated synostosis, several studies have been able to show an elevated incidence of disorders in neuropsychological development. Within this group, concentration and learning difficulties, for example, are particularly common in metopic synostosis, but also occur in sagittal synostosis. Studies have shown that surgery for sagittal synostosis enables children to make up for the previously observable delay in their early development, while children with sagittal synostosis who did not undergo surgery were not able to catch up in terms of development. The effect was noticeable over a long period. Other studies have claimed that the more extensive the surgery that is performed, and the earlier it is carried out, the more favorable the outcome is from a developmental point of view. These studies have had a major impact, but have methodological weaknesses.
Latest results
We have measured full-scale IQ, using the Wechsler Adult Intelligence Scale (WAIS-IV) from 16 years of age and Wechsler Intelligence Scale for Children (WISC-IV) for 6–16-year-olds, in children operated on for craniosynostosis. Our dropout rate is uniquely small, which may be due to the fact that we have called upon patients living in Gothenburg and then expanded the material, step by step, by contacting patients who lived increasingly far away from Sahlgrenska. We have thus avoided nonresponse and dropout due to excessively long journeys. Preliminary results indicate an entirely normal IQ for isolated synostosis, and we have a mean value and a spread of the values that are congruent with the normal distribution. These results deviate from several publications in the field, where mean values for full-scale IQ that are very high, up to 112, are seen — a finding that seems wholly implausible. Either these studies have a large selection bias or the instruments were used incorrectly. Our results indicate that we use the instruments correctly and that our strategy of selecting patients who live a short distance from Sahlgrenska yields a uniquely small selection bias.
A specific test of attention, the Conners Cognitive Performance Test (CPT), has shown several minor deviations from norm data.
General abilities were investigated on the basis of reports from the parents (Adaptive Behavior Assessment System, ABAS), and here too we find minor deviations.
Taken together, these studies show that adolescents who have undergone surgery for sagittal synostosis and metopic synostosis have normal development, but that several minor deviations from the norm are identifiable.
Project group
Peter Tarnow
Sara Fischer